
Overview
Retinitis pigmentosa (RP) comprises a group of inherited retinal dystrophies characterised by the primary degeneration of rod and cone photoreceptors. It is the most common inherited retinal dystrophy with a worldwide prevalence of approximately 1 in 4000.
Over 90 genes have been implicated in RP with more added to the list each year. Autosomal recessive is the most common inheritance pattern account for 50-60% of RP patients, followed by autosomal dominant (30-40%) and X-linked (5-15%).
The initial symptom of RP is reduced night vision and difficulty with dark adaptation, followed by a progressive loss of the visual field in a concentric pattern and eventually loss of central vision. In the ‘classic’ presentation of RP, difficulty with dark adaptation begins in adolescence, and mid-periphery field loss becomes apparent in young adulthood. Typically, when the patient reaches middle age, central cone degeneration leads to a decline in visual acuity. Nevertheless, there is wide heterogeneity among RP patients differing in the age of onset, the severity of visual loss, and the rate of progression.
The hallmark features of RP include bone spicule pigmentation in the mid-periphery, attenuation of retinal vessels, and waxy pallor of the optic nerve head. However, these signs may be subtle or absent in the early stages.
Other associated clinical findings may include epiretinal membrane formation, posterior subcapsular cataract, macular hole formation and cystoid macular oedema. In early-onset RP, nystagmus and high refractive error are also common.
The earliest change on OCT shows disorganisation of outer retina layers, first in the interdigitation zone, followed by the ellipsoid zone, then the external limiting membrane, and finally the RPE. The inner nuclear layer and ganglion cell layers remain relatively well preserved in the early stages of the disease but degenerate later in the disease process.
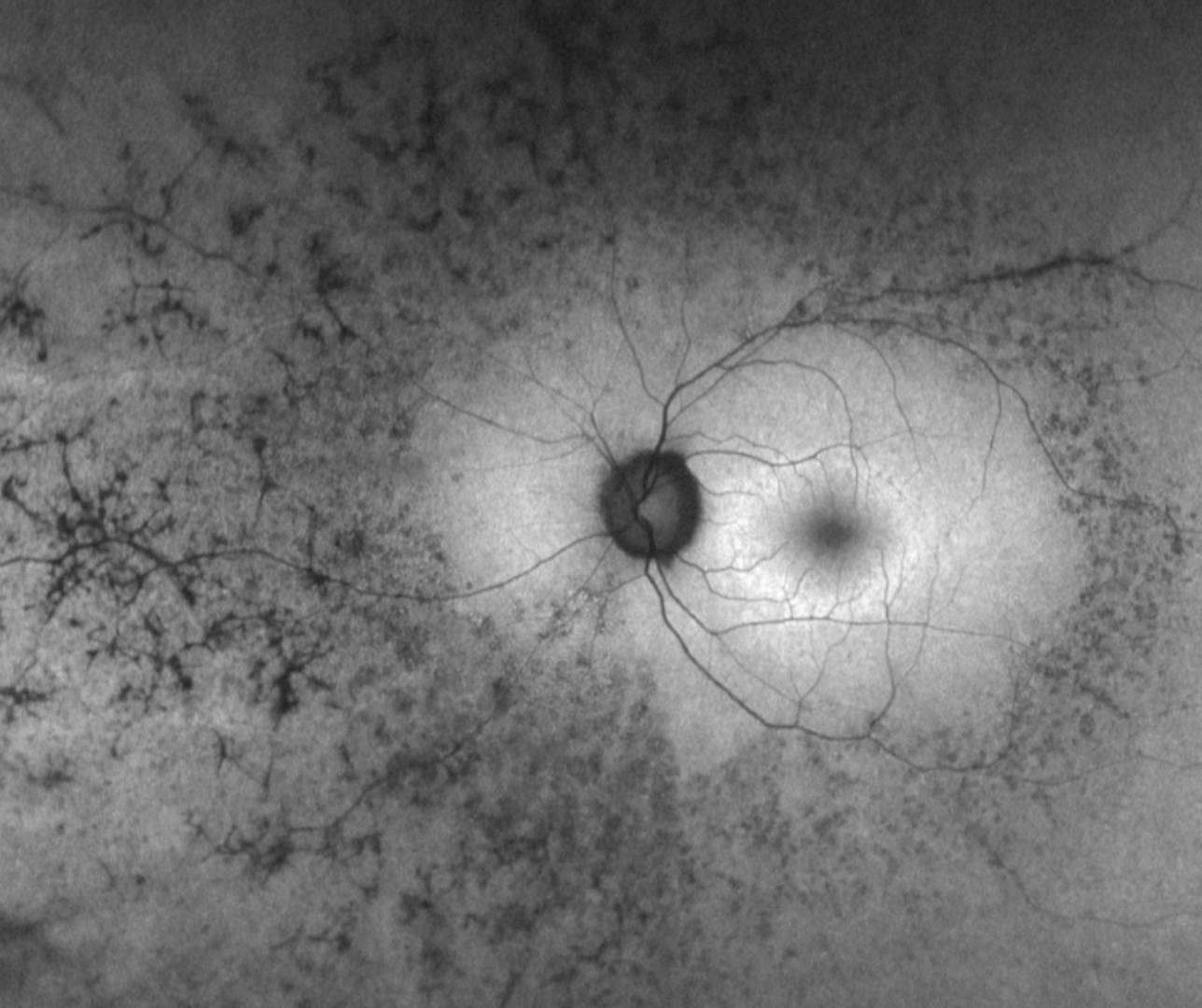
The fundus autofluorescence pattern in RP typically shows hypo-fluorescence in the areas of the bone spicule pigmentation. There is a characteristic hyper-autofluorescent ring around the macula that separates the degenerated retina from the intact cells of the macula. The hyper-autofluorescence is thought to be caused by RPE cells under stress and is an indicator of imminent cell death. Over time, this ring constricts and moves closer to the fovea.
Visual field loss begins with isolated mid-peripheral scotomas, progressing to form a ring scotoma over time.
Colour vision may initially be normal; however, over time, blue-yellow colour vision defects typically evolve.
Full-field electroretinogram helps in the diagnosis, quantification of the severity of the disease, as well as monitoring the progression. It typically shows reduced scotopic responses followed by abnormal photopic responses. As the disease processes, the full-field ERG may become non-recordable despite a residual visual field.
Up to 30% of the patients present with a syndromic form of RP associated with extra-ocular abnormalities. The two most common of these are Usher Syndrome (associated neurosensory deafness) and Bardet Biedl syndrome (childhood obesity, developmental delays and renal abnormalities).
Case Examples
-
Case 1: "Typical" RP
A 36-year-old Caucasian male with poor night vision and best-corrected visual acuity of 6/4.8- (20/15-) in each eye.
Colour fundus photographs (right and left eye)
More infoOptomap (1), red separation (2) and green separation (3) images - right eye
More infoOptomap (1), red separation (2) and green separation (3) images - left eye
More infoFundus autofluorescence imaging
More info24-2 SITA Standard visual field
More infoGoldmann kinetic perimetry
More infoElectrophysiology results
More info -
Case 2: Advanced RP
A 52-year-old Caucasian male with best-corrected visual acuity of 6/15 (20/50) in the right eye and 6/12- (20/40-) in the left. He reports difficulty with night vision and mobility.
Colour fundus photographs (right and left eye)
More infoOptomap (1), red separation (2) and green separation (3) images - right eye
More infoOptomap (1), red separation (2) and green separation (3) images - left eye
More infoFundus autofluorescence imaging (right and left eye)
More infoSpectralis OCT volume and line scans (right macula)
More infoSpectralis OCT volume and line scans (left macula)
More info -
Case 3: Usher syndrome
A 30-year-old Caucasian female with best-corrected visual acuity of 6/12- (20/40-) in the right eye and 6/15 (20/50) in the left.
Colour fundus photographs (right and left eye)
More infoRed free images (right and left eye)
More infoOptomap (1), red separation (2) and green separation (3) images - right eye
More infoOptomap (1), red separation (2) and green separation (3) images - left eye
More infoFundus autofluorescence imaging (FAF)
More infoSpectralis OCT volume and line scan (right eye)
More infoSpectralis OCT volume and line scan (left eye)
More info30-2 SITA standard visual field
More infoElectophysiology results
More info -
Case 4: Unilateral RP
A 48-year-old Indian female with best-corrected visual acuity of 6/7.5 (20/25) in the right eye and 6/6+ (20/20+) in the left. The patient notes difficulty with her left peripheral vision.
Colour fundus photographs
More infoOptomap (1), red separation (2) and green separation (3) images - right eye
More infoOptomap (1), red separation (2) and green separation (3) images - left eye
More infoFundus autofluorescence imaging - FAF (right and left eye)
More infoSpectralis OCT line scans - right (1) and left (2) macula
More infoSpectralis OCT volume (1) and line (2) scan - nasal retina (right eye)
More infoGoldmann kinetic perimetry
More infoElectrophysiology results
More info -
Case 5: Sector RP
A 67-year-old Asian male with best-corrected visual acuity of 6/6 (20/20) in each eye. He has difficulty with night vision.
Optomap (1), red separation (2) and green separation (3) images - right eye
More infoOptomap (1), red separation (2) and green separation (3) images - left eye
More infoFundus autofluorescence images (right and left eye)
More infoSpectralis OCT volume and line scans (right macula)
More infoSpectralis OCT volume and line scans (left macula)
More info
Differential Diagnosis
Other differentials to consider include drug-induced toxicity, chorioretinal infections, sequelae of inflammatory disease and Vitamin A deficiency.
References
Falfoul, Y., Elleuch, I., El Matri, K., Ghali, H., et al. (2020). Multimodal Imaging in Retinitis Pigmentosa: Correlations among Microvascular Changes, Macular Function and Retinal Structure. Journal of current ophthalmology. 32. 170-177.
Phelan, JK., Bok, D. (2000) A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Molecular Vision 2000; 6:116-24.
Sanne K. Verbakel, Ramon A.C. van Huet, Camiel J.F. Boon, Anneke I. den Hollander, Rob W.J. Collin, Caroline C.W. Klaver, Carel B. Hoyng, Ronald Roepman, B. Jeroen Klevering, (2018) Non-syndromic retinitis pigmentosa, Progress in Retinal and Eye Research, Volume 66, Pages 157-186.
Schuerch, K., Marsiglia, M., Lee, W., Tsang, S. H., & Sparrow, J. R. (2016). Multimodal imaging of disease associated pigmentary changes in retinitis pigmentosa. Retina 36 Suppl 1(Suppl 1), S147–S158.